clonalExpansion
Scientific Computing
Dynamic simulation modelling and Bayesian inference for models of clonal Expansion
Mitochondria and Clonal Expansion
What are Mitochondria?
Living things are thing made of cells, the building blocks of life. Cells are very diverse in size and function, but humans are made up of tens of trillions of cells with an average size of 100 micrometers in diameter. Within individual cells, there are smaller sub-units, known as organelles, which have their own specific functions. Mitochondria are a type of organelle, often known as the “powerhouse of the cell” and are responsible for creating the energy required by cells, through the conversion of oxygen and nutrients (sugars, fats and proteins) to adenosine triphosphate (ATP). ATP is the chemical which allows the cell to do many of the jobs it is required to do, through the process of aerobic respiration.
 Eukaryotic cells, including human cells, contain organelles: subunits with specific function. Mitochondria are organelles within eukaryotic cells that specialise in the production of energy.
Eukaryotic cells, including human cells, contain organelles: subunits with specific function. Mitochondria are organelles within eukaryotic cells that specialise in the production of energy.
Mitochondria have not always existed in their current state. The endosymbiotic theory states that mitochondria are descendants of bacteria; free, independent, single-celled, prokaryotic organisms, unlike the mitochondria of today (which are contained within cells). At some point in evolutionary history a predecessor of modern day mitochondria might have been absorbed by the predecessor of a modern day cell, and somehow gave that cell a survival advantage. By the process of natural selection these synergistic partnerships gained a competitive advantage over others resulting in the eukaryotic cells of today generally containing mitochondria.
Mitochondria play an important role beyond just producing energy. Another one of the many processes they are involved with is cell death (apoptosis), a highly important process which is vital for normal function. This page describes the uncontrolled growth of cells which results in tumours and suggests that mitochondria can influence the development of cancers.

A diagram of the inner structure of a mitochondrion
Mitochondria produce over 90% of the energy required by cells to carry out their functions in the body and thus any malfunction of the mitochondria can result in many negative health effects, particularly in tissues that have high energy requirements. Muscle cells are an obvious example and patients with mitochondrial diseases that survive to adulthood can develop severe and debilitating muscle weakness, can become intolerant to exercise and can suffer from fatigue. A classic manifestation of mitochondrial disease is droopy eyelids, where the muscle tissue controlling the eyelids becomes severely dysfunctional.
Mitochondrial diseases are the most common inherited metabolic disorder. Gorman et al. (2016) summarise the various studies on mitochondrial disease prevalence, with on the order of 10 - 100 cases per 100,000 individuals in the North East of England being recorded. In addition to mitochondrial disease, mitochondrial malfunctions have also been linked to a range of disorders, from diabetes to Parkinson’s disease. A better understanding of how mitochondria function is critical for developing treatments for many diseases and improving the quality of life of the many people affected by these diseases.
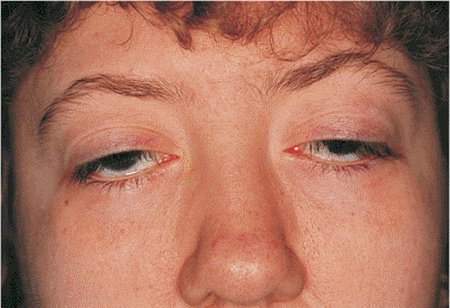
A typical symptom of childhood mitochondrial disease, drooping eyelids
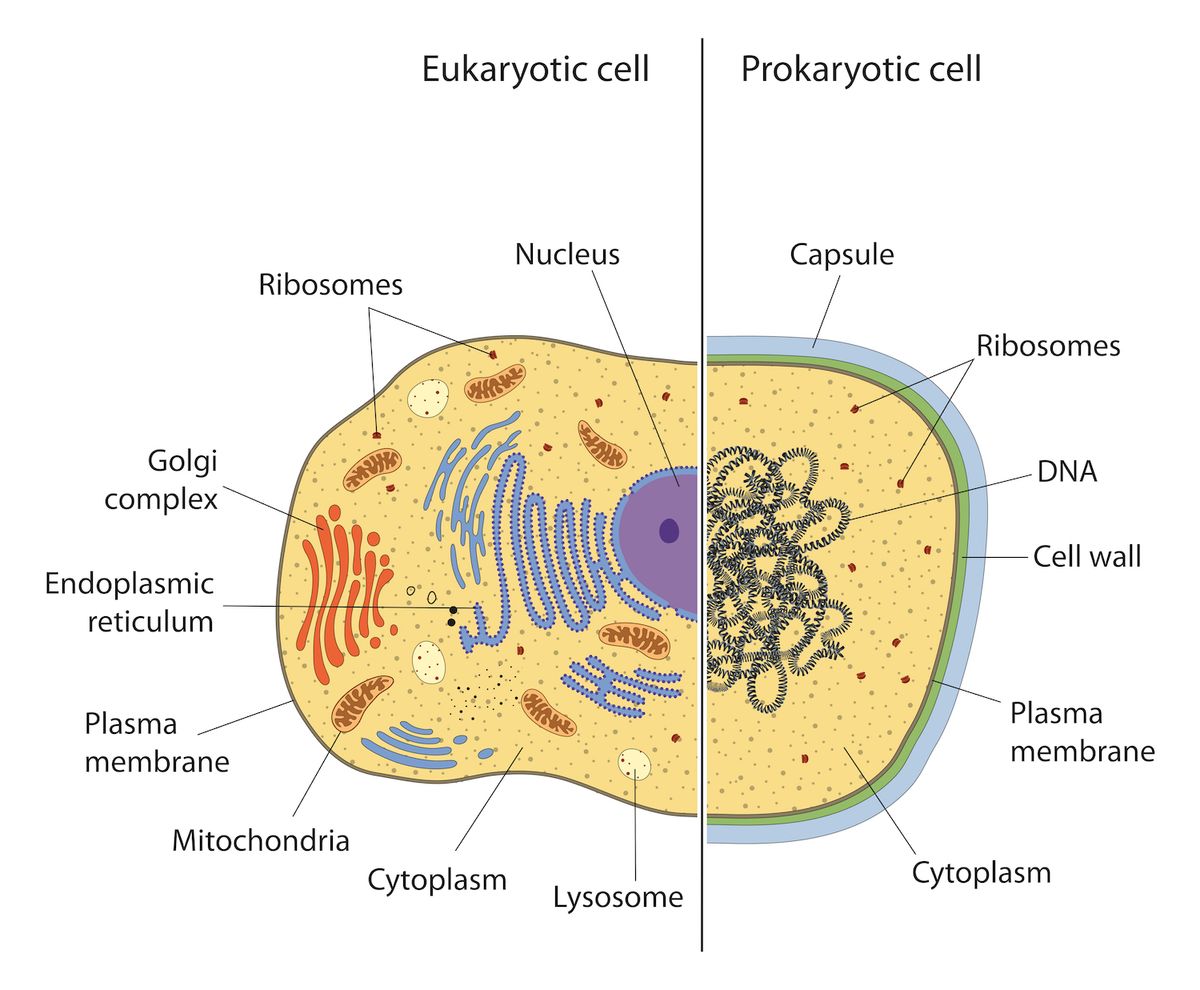
Eukaryotic vs Prokaryotic cells. Only eukaryotic cells contain mitochondria. Multiple copies of circular DNA are found within individual mitochondria, much like they are found within single prokaryotic cells
Mitochondrial DNA
DNA is found in every cell, and contains genetic information which is the instructions for building proteins from which cells are made. The majority of a eukaryotic cell’s genetic material is stored as linear chromosomes in the nucleus, but mitochondria have their own separate, circular genome, known as mitochondrial DNA (mtDNA). DNA can be considered to be the ‘unit of inheritance’, and all nuclear DNA is inherited from both of your parents whereas mtDNA is only inherited maternally.
Unlike the linear structure of nuclear DNA, mtDNA is circular in shape, and far smaller. It shows a lot of similarity to bacterial DNA, and whilst the genome in the nucleus holds several billion base pairs, mtDNA is made up of only 16,569 base pairs. It should be noted that this arises quite naturally from the endosymbiotic theory, as all prokaryotic genomes are circular as well. Nuclear DNA stores about 200,000 times as much information. Another key difference to nuclear DNA is the fact that all mtDNA is inherited from the mother, rather than from both parents. This means we will share mtDNA with siblings, mothers, and any female ancestors on the mother’s side of your family, but none whatsoever with the father’s side.
 Electron microscope image of a slice through two adjacent mitochondria
Electron microscope image of a slice through two adjacent mitochondria
A very important feature of mtDNA is that there are many mitochondrial genomes within a single cell. This is in contrast to the nuclear genome, where there are essentially two copies. Depending on the species and tissue, the number of mitochondria in a cell can vary greatly, from a single mitochondrion per cell all the way up to several thousand. For example, muscle cells typically have lots of mitochondria as muscle cells need to be able to generate energy very quickly when we exert ourselves. Similarly, neurons are also very energy demanding; mitochondrial disease also causes neurodegeneration. Furthermore, each of these individual mitochondria can contain tens of mtDNA, meaning that the overall number of mtDNA in a cell can be in the thousands. It can be as high as ~500,000 per cell in human oocytes, for example.
I will in general refer to two types of mtDNA; the “wild type” mtDNA and the “mutated” mtDNA. Wild type mtDNA molecules refer to those that are widespread and the ordinary type that occur in nature and mutant mtDNA molecules that have been changed in some way, such as through a point mutation or deletion. A point mutation is when one base gets changed, i.e. a change at one of the ~ 16,000 locations in the mtDNA molecule. If this point plays a critical role in the instructions for making a mitochondrial protein, this miniscule change can cause disease. DNA deletions refers to a chunk of the mtDNA circle being removed or ‘deleted’, with the two broken ends being fused back together. For a large-scale deletion a considerable proportion of the ~ 16,000 bases will be missing.

The genetic structure of mitochondrial DNA
Clonal Expansion
Clonal expansion, as described in a review by Lawless et al. (2020), is the process by which populations of mtDNA mutations increase in individual cells. An important theory explaining the progression of some forms of mitochondrial disease is that the effects of disease are strongly linked to clonal expansion of mutations.
As there are multiple mtDNA in an individual cell, the cell can exist in a state of heteroplasmy, meaning that it contains a mixture of wild type and mutated mtDNA species. We can define the mutation load as the proportion of mutated molecules. High mutation loads in individual cells are thought to cause a range of mitochondrial diseases.
In studying clonal expansion we are trying to determine how the levels of mutation load evolve in a range of cells over time. Mutations within a cell could have been inherited from parents, or could arise by random mutation during mtDNA replication, either as part of the disease process or as part of normal aging. The evolution of the system can be thought of as being a discrete, stochastic process. This just means that the population of mtDNA can only take integer values (discrete) and that there is an inherent uncertainty, or randomness (stochasticity) associated with the process. There is strong evidence for stochasticity: a ‘mosaic’ pattern of biochemical deficiency can be seen in the tissues of patients with mitochondrial disease, which suggest that mtDNA dynamics in adjacent cells are independent and random.
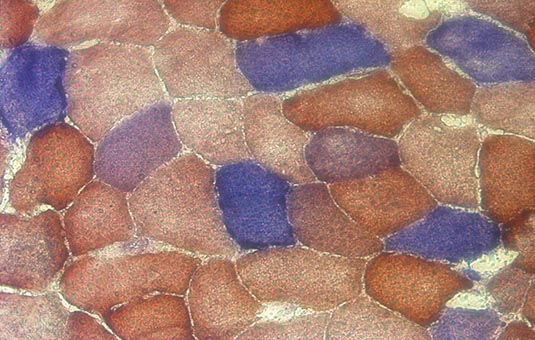
The mosiac pattern as seen above, with the different colours of cell indicating different levels of biochemical deficiency. One theory of mitochondrial disease progression is that the deficiency arises once mutation load becomes high enough.
An area of ongoing research within the scientific community is the question of whether mutated mtDNA may hold some replicative advantage over wildtype mtDNA. A baseline model of ‘random genetic drift’ can be used as a benchmark; random genetic drift just means that none of the mtDNA hold any form of advantage over any other type of mtDNA, meaning in a given cell a mtDNA will be chosen at random to replicate.
Even if mutation events are relatively rare, given that we are made up of lots of cells, each containing many mtDNA, there is a non-negligible chance that high mutation loads could arise in some cells through random genetic drift alone. Elson. et al (2001) showed that random genetic drift in certain cases explain the clonal expansion of mtDNA in individual cells with age. However, there are sill many instances that seemingly cannot be explained by random genetic drift alone, which implies that there might be some sort of selection pressure. For example, one proposed theory stated that mtDNA with a deletion would be slightly smaller and thus replicate more quickly, growing to large populations in this way. Currently, there is no singular accepted theory for the mechanisms or processes by which mtDNA clonally expand that explains all of the observed data.
There are a large range of diseases that can be attributed to malfunctions of the mitochondria; this is unsurprising given the important role that mitochondria play in normal cellular function. Sufferers of mitochondrial disease have a huge decrease in life quality and frequently die at young ages. There are currently no proven treatments of mitochondrial disease and currently the best medicine can offer is an alleviation of some of the symptoms. As previously mentioned, mitochondrial dysfunction has also been seen to be involved in other diseases such diabetes, Parkinson’s and cancer amongst others; the clinical value of a better understanding of clonal expansion is potentially very high.